
Two theoretical polymer science groups at Martin Luther University in Halle investigate the chemically very complex nature of functional polymers, with a wealth of intrinsic and tunable properties. The bandwidth of their micro and macrostructure is just as rich as that of, e.g., proteins in biochemistry. Therefore, a complete understanding of structural and dynamical features is often only possible with the help of computer simulations. Work in Wolfgang Paul’s group is mainly based on Monte-Carlo (MC) simulations and molecular dynamics (MD) simulations from the point of view of theoretical physics. In Daniel Sebastiani’s group ab-initio approaches combine density functional theory (DFT) with MD simulations from the discipline of theoretical chemistry. The very large range of spatial and dynamical effects that are observed in polymer materials requires the synergistic combination of simulation techniques from the atomistic scale up to the mesoscopic scale.
Proton Conduction in Polymer Membrane Fuel Cells
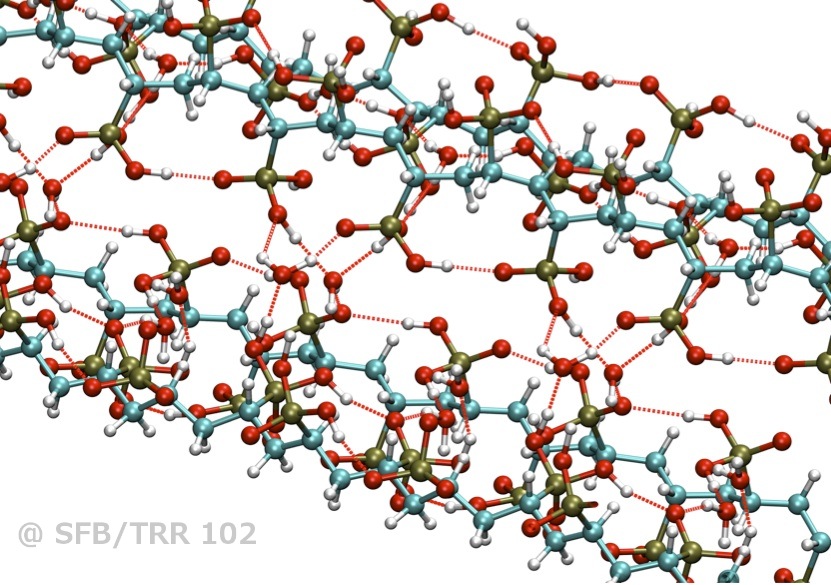
Phosphonic acid-bases polymer chains exhibit a complex hydrogen bond network
The motion of individual protons in functionalized polymers is of great impact for the design of optimized proton exchange fuel cell membrane materials. The most common material (Nafion®) relies on relatively bulky water channels inside a swollen polymer matrix, which restricts its operating conditions. Quantum-chemical molecular dynamics simulations reveal how a derived material (polyethylene functionalized with phosphonic acid) has to be optimized chemically to allow quasi water-free proton conduction at very high operating temperatures (thus saving catalyst material in a fuel cell).
Supramolecular Packing in π-conjugated Polymers
The efficiency of solar cells and organic LEDs can be considerably improved by the help of semiconducting polymers, which increase light harvesting abilities and carrier mobilities in these devices. An important aspect of the functionality of these polymers is their supramolecular packing, which can differ as a function of their processing conditions. The local packing structure, however, is not easy to determine, by means of molecular structure and motion on the nanometer scale. The knowledge of the quantum mechanical orbitals of the electrons, however, enables the calculation of these packing motifs, along with subtle spectroscopic features which allow an experimental confirmation. Most important are NMR chemical shift variations due to hydrogen bonding and supramolecular packing structures, but also red-shifts in fluorescence spectra due to dielectric relaxation effects on femto and pico-second timescales.
Polymer based Composites
Composite materials based on filled polymer matrices have a huge span of possible applications, from relatively low-tech ones like car tires, to high-tech ones like airplane wings. A solid particle dispersion inside the polymer is used to create a composite with superior mechanical properties. A detailed molecular understanding of the interphase region between nano-fillers and polymers, on which the property improvement is based, is still lacking. Chemically realistic molecular dynamics simulations provide such a molecular understanding.
Single Polymer Chains
For bio-polymers like proteins, DNA, and RNA, we are well aware that their specialized and highly evolved bio-chemical functions rely on their spatial structure, which in turn results from the interplay of their energetics determined by their chemical sequence and the one defining feature of all polymers: conformational entropy. But the same holds true for all synthetic polymers as well. Computer simulations via coarse grained models have changed some of the paradigms in this field. Contrary to text book beliefs a flexible chain can indeed exhibit a first-order collapse in presence of a short enough range of attractive interactions. Interestingly, nature has realized this in two-state folding proteins.
[slideshow_deploy id=’92’]
Microphase-separated Diblock Copolymers
Diblock copolymers self-assemble into a variety of microphase-separated structures. This reproducible structure generation on a scale of the gyration radius of the blocks (about 10 nm) is employed for lithographic masks in chip design as well as for bulk hetero junction photovoltaic devices. To model these morphologies, a coarse grained model working on the scale of the radius of gyration is sufficient and a way to reach the extended length and time scales involved in the self-assembly of these morphologies.